定量药理学引导药物开发案例
| 分类 | 亚分类 | 使用方法 | 案例 |
|---|---|---|---|
| 起始剂量估计 | 起始剂量估计 | NOAEL法、MABLE法(本质与PK/PD建模法相同)、PK外推法(异速放大法)、类似药法、PBPK法、IVIVC法(肝代谢引用) | 确定DS8201起始剂量 |
| 剂量相关 | 剂量调整、间隔调整 | PopPK法、PBPK法、PK-PD转化模型、肿瘤生长模型 | Pembrolizumab: 240mg Q2W方案延长至Q6W,或480mg Q4W方案延长至Q8W MCLA-128: 确定Q3W给药一次 |
| 特殊人群剂量调整 | PopPK法 | 万古霉素、多粘菌素,根据肝肾功能调整剂量 | |
| 给药延迟、中断、漏服的影响 | PopPK法、PBPK法 | AC007 根据PK/PD免除II期 | |
| 为后续临床试验确定剂量 | PK-PD模型 | RMC-6236 | |
| RP2D剂量确定 | PK-RO(受体占位)模型、PK-PD转化模型、肿瘤生长模型 | Pembrolizumab MCLA-128 Tepotinib |
|
| 暴露-效应关系 | 暴露-效应关系 | E(暴露量)-R(FT3、FT4等)分析 | MGL-3196 |
NOAEL
未观察到不良反应的剂量,No Observed Adverse Effect Level
MABEL
最小预期生物效应水平,Minimum Anticipated Biological Effect Level
PBPK
生理学的药代动力学,Physiologically Based Pharmacokinetics
IVIVC
体外一体内相关性,In Vitro-In Vivo Correlation
PopPK
群体药代动力学,Population Pharmacokinetics
PK-PD
药代药效学模型,Pharmacokinetics-Pharmacodynamics
E-R
暴露-反应关系,Exposure-Response
定量药理学支持DS8201起始剂量选择
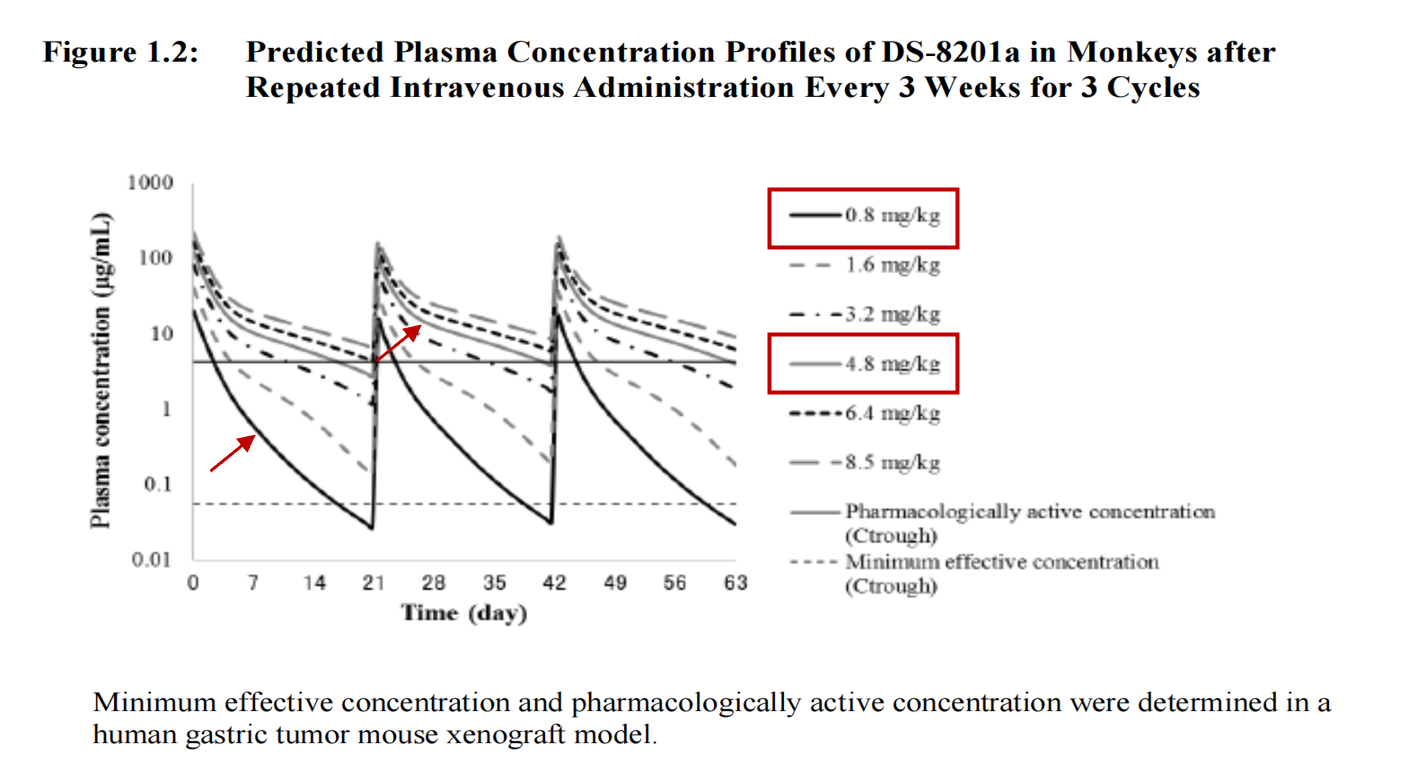
DS8201为已成功上市用于乳腺癌治疗的ADC药物;采用食蟹猴的药代数据模拟人多次给药(Q3W 3cycles)的血药浓度。
- 由临床前暴露-效应关系得出,人体预期的最低起效浓度为0.0551ug/mL,人体药理活性浓度4.26 µg/mL。
- 基于食蟹猴的药代动力学,模拟人体多次给药(Q3W 3cycles)的药时曲线。
- 0.8mg/kg剂量下的谷浓度接近最低起效浓度,故选择0.8mg/kg作为起始剂量。
- 4.8mg/kg剂量下的谷浓度接近药理活性浓度,最终上市剂量为5.4mg/kg,与模型推导剂量基本一致。
定量药理学支持RMC-6236剂量选择
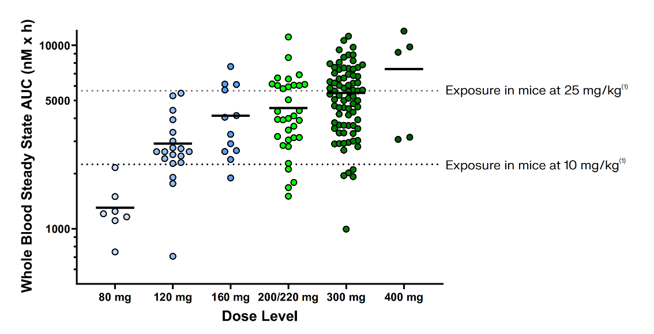
RMC-6236是RAS(ON) 多重选择抑制剂,拟开发治疗晚期胰腺癌;已完成剂量递增和扩展研究,获得各个剂量下单药治疗的药代动力学数据。
- 建立剂量组(横坐标)-暴露量(AUC)的关系。
- 10 mg/kg的RMC-6236可在敏感肿瘤小鼠模型体现出良好的抑瘤率,而25 mg/kg的剂量在大多数肿瘤小鼠模型中可抑制肿瘤生长。将10mg/kg和25 mg/kg剂量下的小鼠暴露量校正后,获得人体对应的药理活性暴露量区间。
- 人体300mg剂量组暴露量绝大部分落在推导的人体对应的药理活性暴露量区间内,支持300mg作为下一阶段临床研究剂量的重要临床药理学证据。